Recently, Liu Juntao et al from School of Mathematics and Statistics, published their latest study entitled "TransBorrow: Genome-guided transcriptome assembly by borrowing assemblies from different assemblers" in the international top journal Genome Research (IF: 11.093). Dr. Yu Ting from Research Center for Mathematics and Interdisciplinary Sciences and Dr. Mu Zengchao from School of Mathematics and Statistics are the co-first authors, and Dr. Liu Juntao is the corresponding author.
High-throughput RNA-seq technology makes it possible to identify and quantify all expressed RNAs from a sample and, therefore, enhances the capability of exploration of complex transcriptomic landscapes. This clearly sheds light on the study of complex diseases related to abnormal splicing events or expression levels, such as cancers. However, it is a highly challenging task to computationally and accurately recover all the expressed transcripts along with their abundances via assembling the huge amount of short RNA-seq reads. Although an increasing number of transcriptome assembling methods have been developed, they are all limited by their accuracy in practical applications. Moreover, no assembling method consistently generates the most accurate assemblies across different kinds of data sets, and it is difficult to determine which assembler to use for a specific data set. Based on above considerations, Liu Juntao et al developed a novel assembly method, TransBorrow, which consistently achieves the best assembly performance on different kinds of data sets by borrowing assemblies from different assembly methods.
As shown in the Figure below, TransBorrow, assembles transcripts by first building splicing graphs based on the mapped reads and extracting reliable paired subpaths from splicing graphs. It then borrows reliable subsequences from different assemblies by building a so-called colored graph. Then, those reliable subsequences and paired subpaths are mapped to the splicing graphs as reliable subpaths for guiding the correct assemblies of expressed transcripts. Finally, a newly designed path extension method is applied to search for a transcript-representing path cover over each splicing graph by seeding those reliable subpaths. Since effectively combining the assemblies from different assembling methods, TransBorrow consistently achieves the best performance of transcriptome assembly across different kinds of data sets.
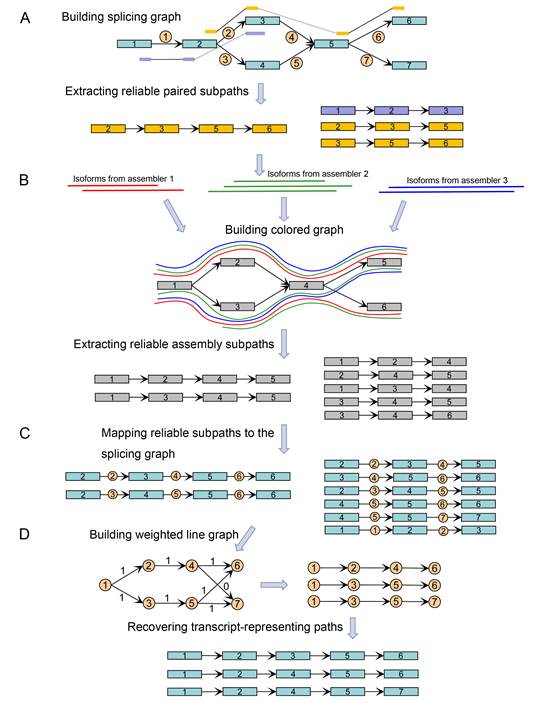
Figure 1. Flowchart of the method TransBorrow
The performance of TransBorrow was tested on more than 100 different kinds of RNA-seq data sets (including both simulated and real data sets). On the simulated data, TransBorrow reaches the highest assembly accuracy among all the compared leading assembling methods including StringTie2,Scallop,Cufflinks,StringTie-merge and TACO. In detail, TransBorrow correctly detected 5.64%-52.29% more expressed transcripts than the other compared assemblers. Then on the real RNA-seq data sets, the advantages TransBorrow is more obvious. E.g., it correctly identified 14.61%-114.93% more expressed transcripts than the other compared assemblers. Moreover, TransBorrow shows more significant superiority in terms of recovering transcripts with low expression levels. For example, TransBorrow correctly detected 7.3%-146.25% more lowly expressed transcripts than the other compared assemblers on the simulated data. And on the real data sets, the improvement achieves 44.19%-361.22%. In addition, TransBorrow also shows much better performance than all the other assemblers in assembling long non-coding RNAs and single-cell RNA-seq data sets.
To the best of our knowledge, TransBorrow is the first genome-guided transcriptome assembler that uses assemblies from different tools by searching for reliable assembly subpaths from different assemblies and then seeding these subpaths for transcript-representing path extensions in each splicing graph. The software has been developed to be user-friendly and is expected to play a crucial role in new discoveries of transcriptome studies using RNA-seq, especially in complicated human diseases related to abnormal splicing events and expression levels, such as cancers.
The link of this paper:
https://genome.cshlp.org/content/early/2020/08/17/gr.257766.119
Written by:Liu Juntao
Edited by:Che Huiqing