Recently, Prof.Liu Zhaopeng’s group of the School of Pharmaceutical Sciences, Shandong University, made great advances in the discovery of potent and water-soluble tubulin polymerization inhibitors that were also effective for the multidrug resistant tumor cells. The results are published onJournal of Medicinal Chemistry,2020,63, 14840–14866, the title is “SAR Investigation and Discovery of Water-Soluble 1-Methyl-1,4-dihydroindeno[1,2-c]pyrazoles as Potent Tubulin Polymerization Inhibitors”.
Tubulin is anα,β-heterodimer that forms the core of the microtubule. Microtubule dynamics are important targets foranticancer drug developments.Microtubule-targeting agents(MTAs), taxol, epothilone and vinblastine, are now widely used in clinic as anticancer drugs whether as a single dosage or in combination with other tumor-targeted drugs or incancer immunotherapy.However, these MTAs suffer from some neurotoxicity andmyelosuppression-related toxicity as well as formulation issuesdue to their low water solubility.In addition, the appearance ofmultidrug resistance also limits their antitumor efficiency. Toreduce the side effects of MTAs and overcome their drugresistance,Prof. Zhao-Peng Liu’s groupapplied the privileged indenopyrazole scaffold in the rational design of the tubulinpolymerization inhibitorstargeting the colchicine binding site, discovered a lead compound,2-(6-ethoxy-3-(3-ethoxyphenylamino)-1-methyl-1,4-dihydroindeno[1,2-c]pyrazol-7-yloxy)acetamide(LL01), that displayed strong inhibition (∼nM) against a variety of tumor cells (J. Med. Chem.2016,59, 5341–5355).Further studies confirmed that LL01 was not a P-gp substrate and effective for multidrug-resistant tumor cells.In HepG2 and KB/V tumor models, LL01 effectively inhibited tumor growth via oral or subcutaneous administration (Investigational New Drugs,2020,38, 29–38;International Journal of Biochemistry and Cell Biology 2017,93, 1–11).Molecular docking simulations indicated that LL01 may have a distinct binding mode with the colchicine site, the 7-acetamide substituent protruded into a new region on the interfacial surface and formed a critical hydrogen bond with Serα178. This unique binding pattern of LL01 may be closely related to its high antitumor activity.
To further improve the water solubility of LL01 (< 5 μg/mL) and to make clear the structureactivity relationships (SARs) of these indenopyrazole analogues,Prof. Liu Zhaopeng group maintained a systematic structural modifications on the 1-methyl-1,4-dihydroindeno-[1,2-c]pyrazole core at the phenolic 6- and 7-positions and the 3-aniline portion. A series of indenopyrazoles were synthesized and evaluated for their tubulin polymerization potency and tumor cell growth inhibitory activity.The SARs supported the predicted binding mode of LL01 and were summarized as follows: (1) an ethoxy group at the indenopyrazole 6-position was the best for high tubulin polymerization inhibition, followed byn-propyl and methyl groups; (2) the substituents at indenopyrazole 7-phenolic hydroxyl should be of proper length and contain a terminal functional group as a hydrogen bond donor to interact withα-tubulin at theα/βinterface, and the preferred substituents were in the order −CH2CONH2> −CH2CH2CH2OH > −CH2CONHOH >−CH2CONHCH3; (3) a substitution at thepara-position ofthe indenopyrazole 3-aniline was not tolerated; and (4) afunctional group, such as an ethoxy, a propionyl, an ethyl ester,and a methyl ester, as a hydrogen bond acceptor at theindenopyrazole 3-anilinemeta-position was favored forinteraction with zone III at the colchicine site, a cyano groupand ethylamine were moderately tolerated, while the carboxylicacid andN-methyl amide were detrimental for the activity; and(5) the replacement ofmeta-ethoxyaniline by 6-ethoxypyridin-2-amine was also well tolerated and favorable for the watersolubility improvement of these indenopyrazoles.
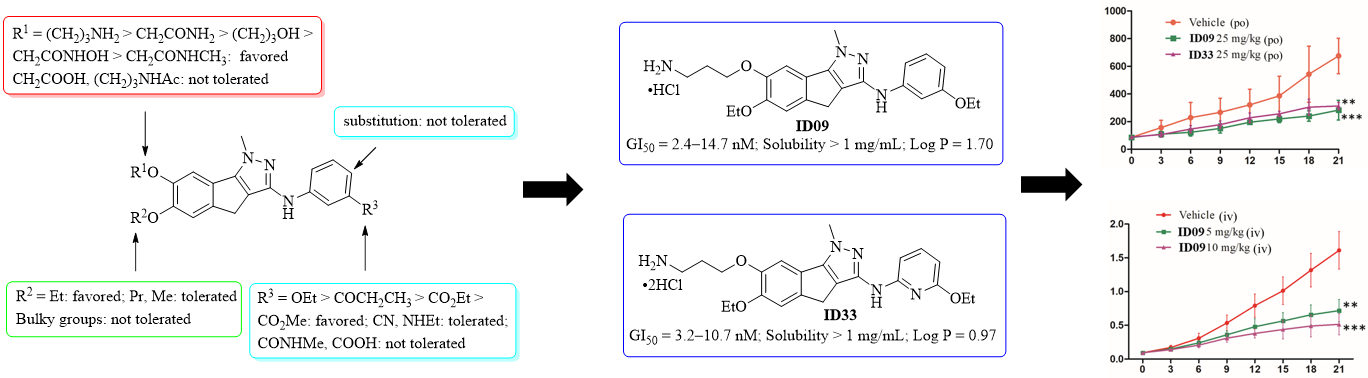
As the hydrochloride salt, the indenopyrazoles ID09 and ID33 targeted the colchicine binding site, inhibited tubulin polymerization and exhibited excellent antiproliferative activity with the GI50values ranging from 2.4 to 14.7 nM.ID09 and ID33 showed lowertoxicity toward the normal liver LO2 cells thanpaclitaxel and CA-4, and were still highly potent against taxol-resistant tumor cells, showing the potential to overcome the multidrug resistance. In HepG2 cells,ID09 and ID33 disrupted cellular microtubule networks, arrested the tumor cell at the G2/M phase and induced the caspase-dependent and mitochondrial apoptosis.At pH 7.4,ID09 and ID33 showed very good and much improved aqueous solubility (>1 mg/mL) than LL01 (>200-fold). They also had lower log P values than that of LL01 and fell into a desirable druglike range (0 < log P < 3). Therefore,ID09 and ID33 have a good balance between their high cellular as well as tubulin potency and the improved aqueous solubility and druglike properties. In a two-week endurance testin normal Kunming mice,ID09 and ID33 were well tolerated, having little effects on the body weights and animal behaviors at least at an oral dose of 50 mg/kg. In HepG2 xenograft mouse models,ID09 and ID33 demonstrated their oral antitumor efficiency at the dose of 25 mg/kg, reducing the tumor growth by more than 50% without obvious signs of toxicity. The iv injection of ID09 at the dose of 10 mg/kg, one time for every other day, resulted in 68% inhibition of tumor growth without obvious toxicity. Therefore,ID09 and ID33 could serve as potential anticancer drug candidates for further developments.
Mr.Cui Yingjie , the Ph.D candidate, and Dr.Liu Chao, the associate research fellow, contributed equally to this work. Dr.Wu Jingde , the associate professor, and Prof. Dr.Liu Zhaopeng were the co-correspondence authors. This work was supported by the National Natural Science Foundation of China (NSFC, Grant No. 81573275), the Key Research and Development Program of Shandong Province (2017CXGC1401).
Links at:https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01345
Written by:Liu Zhaopeng